| | | |
RD-Connect and NeurOmics: mid-year updates |
One of the main achievements of RD-Connect in 2015 has been the launch of a beta-version of the genomics platform as described in our June newsletter. Other aspects of the platform are coming online in stages. The catalogue for patient registries is now accessible and registries are progressively signing up. Biobanks will shortly also be included in the same system, allowing those interested in finding details of what a particular resource contains to quickly find relevant information. Work on a pipeline for transcriptomics and metabolomics is ongoing in collaboration with experts from NeurOmics, EURenOmics and the European Bioinformatics Institute. In collaboration with PhenoTips developers in Toronto, Canada, a PhenoTips instance has been integrated into the RD-Connect platform and will be the primary host of phenotype data for each individual whose genomic data is hosted in the central RD-Connect database. RD-Connect has also brought in three new associated partners over the past 6 months. Each partner is involved in a related area of work which is enabling RD-Connect’s activities to be fully embedded into the wider rare disease and omics research communities. Other highlights to date include: • Publishing the International Charter of principles for sharing bio-specimens and data: the Charter provides guidance for effective legally and ethically grounded sharing and has been published in European Journal of Human Genetics. The Charter has just been submitted for consideration for 'IRDiRC Recommended' status and awaits feedback from the executive committee review board after their meeting this autumn. • Ontology expansion: the Human Phenotype Ontology (HPO) is being used in NeurOmics and EURenOmics to describe and record the clinical features of patients as part of the deep phenotyping in the projects. NeurOmics worked with disease experts, HPO developers and RD-Connect to host an intensive workshop in order to significantly improve the ontology’s coverage of neuromuscular and neurodegenerative diseases. EURenOmics disease experts held a similar event together with the HPO developers to expand coverage of kidney disease phenotypes. This will have significant impact on all those using the HPO for neurological and renal conditions by allowing a more detailed and useful description of clinical features. This in turn will make appropriate matchmaking based on phenotype more likely through tools which use the HPO. • Consenting guidelines: the RD-Patient and Ethics Council have brought together RD-Connect, NeurOmics and EURenOmics in order to provide guidance on the essential elements for informed consent and on procedures to follow when consent is out of date or not sufficient to allow the required use of a sample or associated data. Gaining appropriate consent means that as many as possible of these precious historic sample collections may be used for research. • Catalogues for registries and biobanks, an online searchable catalogue is being developed that provides information about data held by these resources. At present users can access the ID-Card system and propose new registries and biobanks. A data model for the searchable sample-level database for biosamples has been created and is in the process of being tested with data from contributing biobanks. Technical discussions regarding integration of sample-level data with the omics data in the central platform have been held and work on this will progress throughout 2015. |
This month we feature two articles highlighting developments in NeurOmics. The first is from Brunhilde Wirth, Professor of Human Genetics and Head of the Institute of Human Genetics at the Medical Faculty of the University of Cologne in Germany. Brunhilde is leading the work package on identification of novel disease genes in Neurodegenerative (NDD) and neuromuscular (NMD) patients and provides an update on the number of novel genes that have been discovered so far. The second update is from Martin Schulze from the Institute of Medical Genetics and Applied Genomics, Tübingen, who provides an overview of the steps their centre is taking to unravel the genetic causes of hereditary ataxia and paraplegia through the use of disease-specific gene panels and next-generation sequencing. An update on EURenOmics will be featured in a forthcoming newsletter. |
Identification of novel disease genes in NDD/NMD patients A total of 10 NeurOmics partners are searching for novel disease-causing genes in NDD and NMD patients using whole exome sequencing performed at deCODE followed by functional analysis at each academic center. The overall goal is to identify at least 100 novel disease causing genes belonging to the nine disease groups defined within the NeurOmics project. Sequencing data of 595 exomes are available, which is slightly more than half of all samples that will be sequenced by the end of the project. Over 80 % of clinical data has been entered into PhenoTips and consent for data sharing is available. We have identified and already published 51 novel disease genes (41 NeurOmics acknowledged; 10 NeurOmics not acknowledged) all in high ranking journals including Science, Nature Genetics, American Journal of Human Genetics and Brain. A further 40 strong candidate disease genes are in the pipeline and are currently being analysed by NeurOmics partners. A full list of published genes can be downloaded here. ~Brunhilde Wirth, Institute of Human Genetics, University Hospital of Cologne |
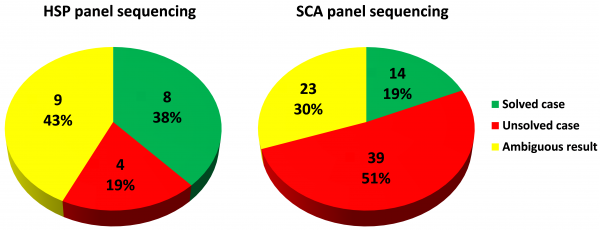
Unraveling the genetic causes of hereditary ataxia and paraplegia using disease specific gene panels and next-generation sequencing Neurodegenerative (NDD) and neuromuscular (NMD) diseases are amongst the most frequent of rare diseases, affecting the life and mobility of more than 500,000 patients and families in Europe. In order to support research, the possibility to develop treatments and improve diagnosis for this large group of patients, the European Commission started the NeurOmics project. NeurOmics aim is to investigate 10 different kinds of rare NDD and NMD with a focus on so-called omics technologies. Hereditary ataxia and spastic paraplegia are among these rare diseases. They are highly heterogeneous and show autosomal dominant, recessive and X-linked forms of inheritance. A cure is still not available and treatment options are thus limited to mitigating symptoms. Many of the patients become wheelchair bound when the disease progresses. The phenotypic spectrum of ataxia and spastic paraplegia is quite broad. Decreased motor abilities are the most prominent but by far not the only clinical features frequently observed. Depending on the genetic background and severity of the disease, patients may also suffer from impaired vision and eye disorders, swallowing difficulties (dysphagia), limited speech abilities (dysarthria), seizures and many other neurological symptoms. Different mutations in different genes are known to cause almost identical phenotypic manifestations of the disease. This makes it challenging for physicians to predict a patient’s disease progression and moreover limits the possibilities of effective genetic family counselling. With the development of next generation sequencing technologies, the possibility of diagnosing a genetic disease by deciphering parts of the patient’s DNA reaches a new level of efficiency, throughput and affordability. The Institute of Medical Genetics and Applied Genomics in Tübingen has almost 8 years of experience in the field of next generation sequencing with a strong focus on diagnosing NDD. For the identification of disease-causing mutations within the NeurOmics project, our aim is to examine 100 patients per disease group using a targeted gene panel approach based on Illumina NGS technology. To enhance the diagnostic yield, we first developed inclusion criteria and selected patients accordingly. As patients may have developed acquired forms of ataxia or paraplegia due to physical trauma, tumors or severe infections of the nervous system, genetic testing wouldn’t lead to any result in these cases and we thus excluded such cases from the study. Moreover, including patients from families with two or more affected raises the probability of a genetic cause of the disease. Finally, we requested the recruiting physicians to exclude more common causes of hereditary ataxia (SCA1,2,3 etc.) and paraplegia (SPG4) prior to panel sequencing. Testing for mutations in distinct hot spots with a relatively high prevalence for the disease at hand is more cost-effective than running an NGS based targeted gene panel. Furthermore, NGS sequencing is not capable of detecting triplet repeat expansion based diseases. This makes a pre-NGS testing for the occurrence of more common triplet repeat expansions a reasonable decision. To support a future genetic diagnosis, an MRI scan should be made available along with a detailed clinical record of each patient. We use PhenoTips, an open source database system, to store standardized and comprehensive clinical data. We established two disease specific gene panels (HaloPlex, Agilent) and kept the content updated on a regular basis as best practice procedure. Our latest disease-specific gene panel covers 181 genes associated with ataxia (hpSCAv8) and 139 genes associated with spastic paraplegia (hpHSPv6), respectively. The underlying target region of 611kb (ataxia) / 391kb (paraplegia) is specifically enriched and sequenced on an Illumina MiSeq device (2 X 150 bp V2 chemistry). The read sequences were analyzed by an in-house bioinformatics pipeline based on BWA and FreeBayes. Currently, index patients from 97 families (ataxia: 76 / paraplegia: 21) are taking part in the study. We achieved a high efficiency with the Agilent HaloPlex platform [target region cov20X: >96% to >97,5% ; mean base coverage: 208±35 (ataxia)/ 366±130 (paraplegia)]. Read-mapping to the human genome followed by annotation using different databases (ClinVar, HGMD, OMIM) resulted in large variant lists: 317-433 (ataxia)/ 145–177 (paraplegia) which were further filtered for rare variants (in-house database, 1000g, esp6500, ExAcc) and for functional relevance. This led to a reduced number of 2-29 variants per patient. Panel sequencing elucidated the molecular basis in 14 out of 76 panel sequenced ataxia families (19%) and 8 out of 21 sequenced paraplegia families (38%) respectively. For both disease groups, potential disease causing variants of yet uncertain significance could be identified in another 35-50% of cases. |
Figure 1 - Results of panel sequencing 1. The figure shows the outcome of diagnosing hereditary ataxia (SCA, n = 76 patients) and spastic paraplegia (HSP, n = 21 patients) using targeted next generation sequencing gene panels. For patients with an ambiguous result, variants of uncertain significance were found which needs further investigation (clinical feedback of attending physician or even functional analysis). These quite convincing initial results prove the project’s inclusion criteria to be appropriate for both, research and diagnostic approaches. The relatively high proportion of ambiguous cases clearly shows the need of further investigations, like segregation analysis of candidate genes. Nevertheless, we could show that panel sequencing is a sensitive, fast and cost-efficient method to identify putative disease-causing mutations in a large number of cases. Within the NeurOmics project, for patients in whom no disease-causing mutation could be detected by panel sequencing, whole exome or genome sequencing will be added on a research basis. This should lead to the discovery of new disease genes which may be included into future panel versions and moreover expand the genotype/phenotype correlation for these rare neurodegenerative diseases. The SCA and HSP panel gene list can be downloaded here. ~ Martin Schulze, the Institute of Medical Genetics and Applied Genomics, Tübingen |
Publications Mutations in GMPPB cause congenital myasthenic syndrome and bridge myasthenic disorders with dystroglycanopathies Belaya K, et al., (2015) Brain [Epub ahead of print] Gene discovery and development of novel diagnostics and therapies for congenital myasthenic syndromes (CMS) is one of the focus areas of NeurOmics. In this recent paper in Brain, the result of a collaboration between NeurOmics partners Newcastle University and University College London with CMS experts at Oxford University, the authors describe mutations in GMPPB as a cause of CMS. CMS are a group of inherited disorders that arise from impaired signal transmission at the neuromuscular junction. Muscle weakness is common in CMS, but highly variable from hour to hour, day to day or month to month. Mutations in at least 20 genes are known to lead to the onset of CMS. Four of these, ALG2, ALG14, DPAGT1 and GFPT1, are involved in glycosylation. There remain a number of CMS cases that have a clear neuromuscular transmission defect, but no identified mutation in the known CMS-associated genes. In this paper, whole exome sequencing is used to identify a fifth glycosylation gene, GDP-mannose pyrophosphorylase B (GMPPB), where mutations cause CMS. CMS due to GMPPB mutations may frequently remain undiagnosed due to lack of facial features usually associated with myasthenia, the presence of high creatine kinase levels and the restricted muscle groups that show decrement on repetitive nerve stimulation. Recognition of this condition is important as patients respond symptomatically to appropriate medication and their quality of life can be significantly improved by appropriate treatment. SIL1-related Marinesco-Sjoegren syndrome (MSS) with associated motor neuronopathy and bradykinetic movement disorder Byrne S et al., (2015) Neuromuscular Disorders 25, 585-8 Marinesco–Sjoegren syndrome (MSS) is a recessively inherited multisystem disorder caused by mutations in SIL1 and characterized by cerebellar atrophy with ataxia, cataracts, a skeletal muscle myopathy, and variable degrees of developmental delay. In this paper, the authors presemnt a case study of SIL1-related MSS, with an associated motor neuronopathy and a bradykinetic movement disorder preceding the onset of ataxia. This case expands the current spectrum of SIL1-related MSS and suggests a continuum with other early-onset multisystem disorders with overlapping clinico-pathological features and putatively linked in the same cellular pathways. Whole exome sequencing and analysis of the patient was performed within the NeurOmics project. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Trautmann A et al., (2015). Clinical Journal of the American Society of Nephrology 10, 592-600. Steroid-resistant nephrotic syndrome (SRNS) is a rare kidney disease involving either immune-mediated or genetic alterations of podocyte structure and function. The rare nature, heterogeneity, and slow evolution of the disorder are major obstacles to systematic genotype-phenotype, intervention, and outcome studies, hampering the development of evidence-based diagnostic and therapeutic concepts. Within EURenOmics, Prof. Corinne Antignac and EURenOmics coordinator, Franz Schaefer lead a dedicated work package which strives to: (1) identify and characterize novel disease-causing and modifier genes underlying SRNS; (2) develop and validate new disease ontology for SRNS; (3) develop and implement rapid diagnostic tools and biomarkers for prognosis; and (4) develop novel molecular therapies for genetic SRNS. Furthermore, the European research consortium, PodoNet, has been formed to investigate diseases affecting the podocyte. The consortium’s goal is to advance understanding of the demographics, causes and prognosis of steroid resistant nephrotic syndrome in children and adults, provide an evidence base for clinical management and explore innovative treatment strategies for genetically based disease. To achieve this goal, the PodoNet Consortium has created an international registry for congenital nephrotic syndrome and childhood-onset steroid-resistant nephrotic syndrome. Below we highlight two papers which have recently been published in collaboration with EURenOmics and the PodoNet Consortium. This recent publication by Trautmann et al., (2015) provides an overview of the PodNet registry cohort which has been collecting data on SRNS. In this paper a summary of this data including age of onset, proportion of individuals with reported extrarenal abnormalities, most common histopathologic diagnosis and most common gene mutations. The PodoNet cohort may serve as a source of reference for future clinical and genetic research in this rare but significant kidney disease. ADCK4-Associated Glomerulopathy Causes Adolescence-Onset FSGS. Korkmaz E et al., (2015). Clinical Journal of the American Society of Nephrology (Epub ahead of print) In this high impact publication by Korkmaz et al., (2015), the authors describe one of the first treatable mitochondriopathies in nephrology. 26 patients from 12 families with recessive mutations in ADCK4 are reported. Patients with ADCK4 mutations showed a largely renal-limited phenotype, with three subjects exhibiting occasional seizures, one subject exhibiting mild mental retardation, and one subject exhibiting retinitis pigmentosa. The paper describes how ADCK4-related glomerulopathy is an important novel differential diagnosis in adolescents with SRNS/FSGS and/or CKD of unknown origin. Full text of all publications available here: Belaya K, Rodríguez Cruz PM, Liu WW, Maxwell S, McGowan S, Farrugia ME, Petty R, Walls TJ, Sedghi M, Basiri K, Yue WW, Sarkozy A, Bertoli M, Pitt M, Kennett R, Schaefer A, Bushby K, Parton M, Lochmüller H, Palace J, Muntoni F and Beeson D (2015). Mutations in GMPPB cause congenital myasthenic syndrome and bridge myasthenic disorders with dystroglycanopathies. Brain [Epub ahead of print]. Byrne S, Dlamini N, Lumsden D, Pitt M, Zaharieva I, Muntoni F, King A, Robert L and Jungbluth H (2105). SIL1-related Marinesco-Sjoegren syndrome (MSS) with associated motor neuronopathy and bradykinetic movement disorder. Neuromuscular Disorders 25, 585-8. Trautmann A, Bodria M, Ozaltin F, Gheisari A, Melk A, Azocar M, Anarat A, Caliskan S, Emma F, Gellermann J, Oh J, Baskin E, Ksiazek J, Remuzzi G, Erdogan O, Akman S, Dusek J, Davitaia T, Özkaya O, Papachristou F, Firszt-Adamczyk A, Urasinski T, Testa S, Krmar RT, Hyla-Klekot L, Pasini A, Özcakar ZB, Sallay P, Cakar N, Galanti M, Terzic J, Aoun B, Caldas Afonso A, Szymanik-Grzelak H, Lipska BS, Schnaidt S, Schaefer F; PodoNet Consortium (2015). Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clinical Journal of the American Society of Nephrology 10, 592-600. Korkmaz E, Lipska-Ziętkiewicz BS, Boyer O, Gribouval O, Fourrage C, Tabatabaei M, Schnaidt S, Gucer S, Kaymaz F, Arici M, Dinckan A, Mir S, Bayazit AK, Emre S, Balat A, Rees L, Shroff R, Bergmann C, Mourani C, Antignac C, Ozaltin F, Schaefer F; PodoNet Consortium (2015). ADCK4-Associated Glomerulopathy Causes Adolescence-Onset FSGS. Clinical Journal of the American Society of Nephrology (Epub ahead of print). |
RD-Connect Registry and Biobank Meeting (BYOD2) From 30 June to 2 July, RD-Connect Registries work package leader Domenica Taruscio and her team at ISS hosted the second “bring your own data” (BYOD) meeting in Rome. This meeting welcomed registry owners, biobank owners and data linkage experts with the main aim of the meeting to move closer towards the goal of linking data from these varied sources. The meeting started with an update from Sabina Gainotti and Paola Torreri (ISS) on ID-Cards, the online searchable catalogue of patient registries. In addition to the phenotypic and genotypic data collected, this system also includes detailed data on the standards, structure and governance of each registry. All registry owners are encouraged to provide as much information as possible to help make this system a valuable resource for the community. Currently the information on the number of cases recorded in registries is uploaded manually by registry owners inside the "disease matrix" and one main aim of the workshop was defining a standardised procedure for the automatic update of the disease matrix through application programming interfaces (APIs). Robert Reihs from the University of Graz is defining the procedure for registries participating in the ID-Card system and there was a lot of discussion among IT experts around the best API solutions. |
Marc Hanauer (Orphanet) presented the work Orphanet has been doing to match rare disease terms with Human Phenotype Ontology (HPO). Great progress has been made, with 2050 diseases annotated to 4500 HPO terms; the total process is expected to be completed by the end of the year. This annotation/ontological theme was carried throughout the two days with an emphasis on the need for registries to consider how they are collecting and coding data in order to make it easier for them to attain interoperability in the future. Agata Robertson (Newcastle) presented an update on the RD-Connect plans for a unique identifier for individuals whose data is held within participating registries, biobanks and the RD-Connect omics platform. To be known as the RD-ID, this aims to ensure it is possible to establish whether particular data items belong to the same individual without identifying the individual or requiring any personal information to be disclosed. This enables data linkage and prevents duplication of data, while protecting patient confidentiality. The system proposed will be unique to RD-Connect but use data items that are interoperable with the HD-ID and ongoing NIH projects. This work is in progress, and the registries in the Core Implementation Group (CIG) are encouraged to see if they collect the appropriate items and if not to consider the resources needed to collect these both prospectively and retrospectively. |
Over the remainder of the meeting Marco Roos (Leiden) led a number of breakout sessions focusing on the various hurdles facing the linking of biobanks and registries. These covered aspects that are important from both the user perspective, e.g. core data elements and a glossary of terms and the technical point of view e.g. data access, data flow and APIs. One group focused on the work Manuel Posada and Estrella Lopez (ISCII) have been doing surrounding common data elements. A clear list of 35 elements has been produced and discussions surrounded what incentive there should be for registries to collect these elements and what, if anything should be mandatory. Again it will be important for the CIG registries to assess the data currently collected and if this could be adapted in the future. The data linkage experts used test data exports provided by the European Huntington Disease Network, Global FKRP Registry and UK FSHD Registry in an excel format to develop a proof of concept. This allowed one group, led by Mark Wilkinson (Madrid) to establish a first draft using a FAIR assessor on top of these exports which uses an API linked data platform. The data linkage experts will develop this method further and test its usability on the registries and biobanks involved in the CIG. It became clear that the expertise for the technical solutions are available within RD-Connect, however the challenges remain in the access to data on a governance level e.g. are the right consents in place and the need to translate these achievements into lay language for users to understand. |
In summary, the outcome of the meeting was very positive and concluded that it will be possible to link all of the data on some level. However, it is now important for registries and biobanks to consider how they need to adapt to these changing technologies and for RD-Connect to consider how to provide incentives for them to do so. This work is ongoing and a follow up meeting will take place in Rome, 24-25 September, the RD-Connect Workshop on Data linkage and ontologies. This follows the 3rd International Summer School on Rare Disease and Orphan Drug Registries. A full meeting report will be available soon. ~ Libby Wood, Curator of the UK Myotonic Dystrophy and FSHD Registry Coordinator |
Sign up for the BioSB RNA-seq data analysis course! Registration is now open for the BioSB RNA- seq data analysis course (7-9 September 2015) which is being coordinated by RD-Connect partner Peter-Bram ’t Hoen (Leiden University Medical Center, The Netherlands) and ErasmusMC. Participants for the RNA-seq course should preferably have participated in the general Next Generation Sequencing (NGS) course or otherwise have demonstrated hands-on experience with NGS data analysis. This advanced course for people with experience in will consist of seminars and hands-on command line, Galaxy and R practicals, and will cover the analysis pipelines for differential transcript expression and variant calling. Examples will be taken from human and mouse studies. The course does not cover prokaryotic RNA profiling nor plant- and metagenomics aspects. Registrations will be handled on a first-come-first-served basis but experience and motivation will be reviewed. Seats have been reserved for participants in the FP7-funded project RD-Connect and Marie Curie European Training Network TranCYST. |
Upcoming events For further information on future events please visit the events page on the RD-Connect website. |
The Human Genetics Society of Australasia 2015 Annual Scientific Meeting, Perth, Western Australia, 8-11th August The HGSA is the foremost scientific body for those working in the field of Human Genetics throughout Australia and New Zealand. Our membership currently stands at over 1000 members in Australasia. The society also supports a number of special interest groups including: • Australasian Association of Clinical Geneticists (AACG) • Australasian Society of Genetic Counsellors (ASGC) • Australasian Society for Inborn Errors of Metabolism (ASIEM) • Australasian Society of Cytogeneticists (ASoC) • Molecular Genetics Society of Australasia (MGSA) Confirmed speakers include RD-Connect partner, Hugh Dawkins and NeurOmics partner, Nigel Laing Further information is available here The 3rd International summer school on rare disease and orphan drug registries Istituto Superiore di Sanità, Rome, Italy, 21-23rd September RD-Connect partners at theIstituto Superiore di Sanità, Rome, Italy will be hostingan International Summer School which will be focused on the specific aims and needs of registries oriented to clinical research, comprising the study of the natural history of diseases, the assessment of treatment effectiveness and post-marketing surveillance of orphan drugs. The School will train participants on the methodologies and resources available for the establishment of a clinical research registry and on the implementation of successful strategies to ensure long time sustainability of the registry, including data sharing and dissemination activities. The Workshop will consist of brief frontal presentations and practical working groups where participants will learn to make their data interoperable with other sources and databases. The working groups will get together registry owners and bio-informatics experts. This event is open to health professionals, researchers, medical specialists, medical students and representatives of patient associations, who are involved or intend to establish a rare disease patient registry. A selection process will apply based on the participant’s background and role with reference to registry activities. Further information is available here: EMBO Workshop - Molecular Mechanisms of muscle growth and wasting in health and disease, 20 September-25 September 2015, Congressi Stefano Franscini, Monte Verità, Ascona, Switzerland This meeting will focus on the molecular mechanisms involved in muscle wasting diseases including cachexia, sarcopenia and muscular dystrophies. Its focus on disease aspects in skeletal muscle, its interactive format and its small size makes this meeting unique. The conference will take place on September 20-25, 2015 at the Conference Centre Monte Verità, Ascona, Switzerland, the venue of choice for Congressi Stefano Franscini, the international conference platform of ETH Zurich. The conference is limited to 120 participants. As we expect more applicants, we highly encourage to submit an abstract, which will help us to select participants and speakers of short talks. Deadline for abstract submission is Friday 12. June, 2015 Further information is available here. 3rd Ottawa International Conference on Neuromuscular Biology, Disease and Therapy, September 24-26, 2015, Ottawa, Canada The CNMD is hosting the 3rd Ottawa International Conference on Neuromuscular Biology, Disease and Therapy on September 24-26, 2015. After two previous successful neuromuscular disease conferences in Ottawa, the 2015 conference promises to offer an outstanding program emphasizing recent breakthroughs in basic and translational research and clinical discoveries in neuromuscular disease. The Conference is structured for both basic researchers and clinicians and will feature internationally-recognized invited speakers highlighting advances in all aspects of NMD research, including novel techniques to diagnose NMD, biology of disease pathogenesis, expanding clinical phenotypes, basic muscle and stem cell biology, and promising therapies to treat these devastating disorders. As in past years, trainees are encouraged to attend and participate – selected abstracts will be featured for platform presentation during the scientific sessions, and all posters are eligible for top poster awards. Confirmed speakers include RD-Connect coordinator Hanns Lochmüller, RD-Connect Associated partner Kym Boycott and NeurOmics infrastructure workpackage leader Volker Straub Further information is available here. BioMedBridges Symposium: open bridges for life science data, 17-18 November 2015, Wellcome Genome Campus, Hinxton, UK The symposium Open bridges for life-science data – to be held on 17-18 November 2015 on the Wellcome Trust Genome Campus in Hinxton near Cambridge, UK – will provide an opportunity for the big – and growing – community to discuss real-life challenges connected to data sharing and interoperability in the life sciences. Topics covered include semantic technologies, translational research infrastructure, APIs and workflows, ethics and security requirements for sensitive data, and metagenomics. Conference workshops include a session for researchers on journal data policies, in which the Scientific Data team is participating. The symposium provides the perfect opportunity to network, discuss and collaborate with other like-minded individuals, including leaders in the field. RD-Connect Impact workpackage leader, Kate Bushby, will be taking part in the podium discussion on Omics and translational research. Furthermore, there will be an RD-Connect workshop available for attendees. Details to follow. Further information available here. TREAT-NMD Alliance Bi-Annual Translational Sciences Conference. December 6 - December 8, Washington D.C., USA This conference will be a fantastic opportunity for patients, academics, clinicians, patient registry curators and industry representatives, to get together to network, learn and exchange ideas about translational research. RD-Connect will be speaking about the role of neuromuscular registries in -omics research and the wider RD-Connect project. Further information is available here |
Why did I get this email? You received this email because you are associated with RD-Connect, EURenOmics or NeurOmics or because you signed up online. We will send out around one email per month with news of relevance to these projects and to IRDiRC. If you don't want to receive any further newsletters, you can unsubscribe using the link below. If you're reading this online or if it was forwarded by a friend, you can sign up to future editions here. |
| | | | | |